image:
Proactive genome assembly (PIGA) with pangenome knowledge
vision Again
Credit: Jian Yang Lab at Westlake University
The research team led by Zhen-Xing He was Professor Jian Yang at the School of Life Sciences, Westlake University, together with colleagues, published their latest findings in Nature on April 1. This study introduced a new method of gene assembly (PIGA). By combining an expensive hybrid sequencing strategy of long and short reads, the team succeeded in constructing a genome for more than a thousand individuals. This achievement overcomes the limitations of previous small sample genomes, and provides a fundamental basis for genetic and clinical research.
Since the completion of the Human Genome Project, single-lineage genes (such as GRCh38) have served as the basis of biomedical research. However, the genetic makeup of individuals varies greatly, and a single reference genome cannot capture much of the genetic diversity across populations. This leads to complex genetic variants, such as structural variants (SVs) and tandem repeats (TRs), which are overlooked in traditional analyses. To address this challenge, researchers proposed the concept of the pangenome—a collection of genetic sequences that represent the genetic diversity of a population.
Although long-term sequencing advances have enabled the collection of high-quality diploid genomes, the high cost of sequencing has limited the sample size of past pangenomes to only a few individuals. Such small datasets are not sufficient to accurately estimate the frequency of genetic variants in a population or to resolve variants of low frequencies and highly complex regions. Therefore, developing a low-cost genome-wide construction strategy for large populations has become an urgent need to address the impact of complex multivariate processes and improve clinical diagnosis.
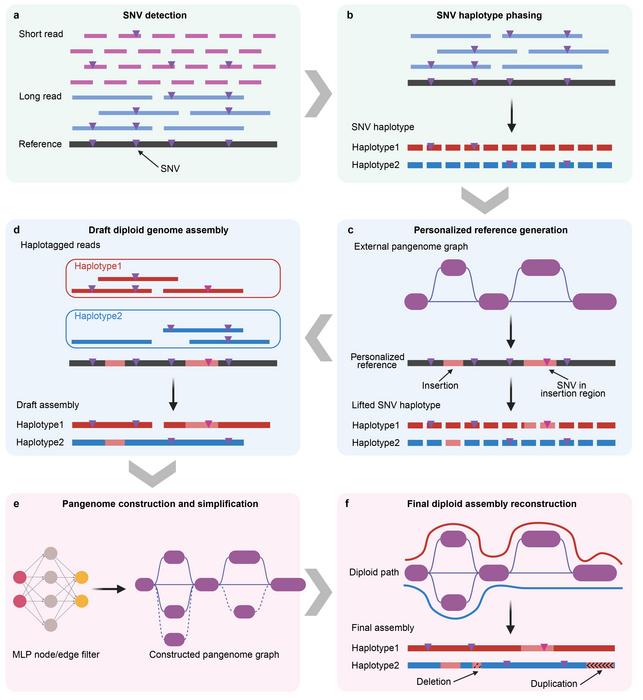
Yang’s group has been devoted to the research of statistical methods of genetics, genomics, and large-scale data analysis of complex human systems. By developing advanced computational methods, the team has tackled fundamental challenges in processing large-scale genomic data. Analysis tools developed by the group, such as GCTA-GREML, SMR, and gsMap, have been adopted worldwide. In order to address the challenge of building large pangenomes, the research group developed a pangenome-informed genome assembly (PIGA) workflow (Figure 1). Unlike de novo assembly methods, which rely on sequencing data from individual samples, PIGA takes a pangenome-guided approach to integrate sequence information across the entire cluster. It fully utilizes a cost-effective hybrid sequencing strategy based on extensive Illumina data and PacBio long-term whole genome sequencing (WGS) data. This approach greatly reduces the cost of sequencing while facilitating the collection of genes from general data, thus providing a new technical avenue for future population sequencing studies.
Using this method, the research team built the world’s largest human pangenome to date, consisting of 1,116 diploid genomes with a quality-quantity (QV) value of 46. Pangenome identified 405.3 million base pairs (Mb) of non-reference sequence missing from current references and GRCh38). Importantly, the team defined 26.2 Mb of these sequences as functional and predicted regulatory elements, greatly expanding our understanding of non-reference sequences in the human genome.
Figure 1. Overview of the pangenome-informed genome assembly (PIGA) process.
By using large amounts of assembly data, the researchers compiled a complete list of genetic variants. In addition to 35.4 million subspecies, this list captured a large number of complex variants, including 110,530 SVs, 485,575 TRs, and 0.86 million nested variants embedded in non-reference sequences.
Using this list, the team identified a variety of medically relevant genes at multiple scales (Figure 2), including mutating SVs, pathogenic TR expansions, different gene clusters, and HLA genes. These findings indicate that the list of 1KCP types provides a valuable reference for the clinical diagnosis of pathogenic mutations.
By combining gene expression data, the team mapped pan-variant expression quantitative trait loci (eQTL). They identified 3,256 eQTLs involving complex variants (SVs, TRs, and nested variants), elucidating the regulatory complexity of these variants.
Together, this study greatly improves our understanding of the complex nature of genes and their functional consequences, creating a new paradigm for human health research and panjinome studies in other species.
Ph.D. student Yifei Wang and Research Assistant Professor Zhongqu Duan are the first authors of the study. Professor Jian Yang is the final author. This work was supported by the National Natural Science Foundation of China, the National Key R&D Program, the “Pioneer & Leading Goose” Program of Zhejiang, and the New Cornerstone Science Foundation. Computer facilities are provided by the Advanced Computing Center at Westlake University.
Professor Jian Yang’s research group aims to develop statistical genetics and bioinformatics methods. By analyzing in-depth genomic and multi-omic data from large population groups, they aim to uncover the genetic architecture and molecular mechanisms underlying complex diseases, translating these discoveries into new diagnostic strategies, drug target discovery, and precision medicine.
Related links:
Link to paper: https://www.nature.com/articles/s41586-026-10315-y
Jian Yang lab website: https://yanglab.westlake.edu.cn/
Research Methodology
Meta-analysis
Research Topic
It doesn’t work
Article Title
China’s 1000 pangenome empowers medical and population genetics
Publication Date of Articles
1-Apr-2026
Description: AAAS and EurekAlert! are not responsible for the accuracy of the information published on EurekAlert! by participating in organizations or for the use of any information through the EurekAlert system.
#Collecting #human #genomes #costeffective #method #powers #future #medicine